국내에서 분리한 딸기누른오갈바이러스의 유전적 다양성
Genetic Diversity of Two Isolates in Strawberry mild yellow edge virus from Korea
Article information
Abstract
Two isolates of Strawberry mild yellow edge virus were newly isolated in strawberry (Fragaria x ananassa) cultivar Selhyang and Kamhong from Korea. The complete nucleotide sequence of the coat protein (CP) of two Korean isolates were determined and analyzed. Sequence identity of nucleotide and amino acid between SH and KH isolates was 90.4% and 95.5%, respectively. The comparison of three Korean isolates including previously reported KNS1 with 45 SMYEV sequences from other countries deposited in GenBank database revealed an identity ranging from 81.2% to 100%. The phylogenetic analysis of CP of all SMYEV isolates showed the five subgroups (I-V), with Korean isolates being divided into two different subgroups. The isolates KH and KNS1 were included in subgroup I, whereas SH was included in subgroup IV which is new phylogenetic subgroup. Genetic diversity analysis indicated that new subgroup had greater variability and nucleotide diversity between subgroups resulted in values ranging from 0.0863 to 0.18004. This report represents the first molecular characterization of SMYEV isolates from Korea.
서 론
딸기누른오갈바이러스(Strawberry mild yellow edge virus, SMYEV)는 1922년에 미국 캘리포니아에서 처음으로 보고된 이후 현재 전세계에서 가장 널리 발병하고 있는 딸기 바이러스 중 하나이다(Horne, 1922; Jelkmann 등, 1990; Martin과 Tzanetakis, 2006; Thompson과 Jelkmann, 2004; Tzanetakis와 Martin, 2013). SMYEV는 분류학적으로 Alphaflexiviridae과의 Potexvirus 속에 속하며, 유일하게 진딧물에 의해 전염하는 Potexvirus로 딸기진딧물인 Chaetosiphon fragaefolii에 의해 영속 전염된다(Jelkmann 등, 1990). SMYEV의 감염증상은 딸기의 품종 및 바이러스 변이주(strain)에 따라 병징 및 피해 정도가 다르다고 알려져 있다. Martin과 Tzanetakis (2006)의 보고에 따르면 감수성 품종의 경우 잎 뒤틀림이나 위축증상 및 잎 가장자리가 황화되는 증상을 보이지만 내성 품종의 경우 전혀 병징이 나타나지 않기도 해 수확량 및 생육 이상과 같은 피해율도 0-30%로 매우 상이하게 나타난다. 국내의 경우 2009년과 2011년에 딸기에서 SMYEV가 보고된 바 있는데, 딸기 품종에 따라 잎 가장자리에 주름 또는 황화, 잎에 모자이크 증상이 나타나지만 무증상에서도 바이러스가 검출되어 바이러스 감염과 병징의 연관성은 적은 것으로 보고하였다(Cho 등, 2011; Choi 등, 2009).
SMYEV는 최초 발생 당시에는 구형바이러스인 Luteovirus로 분류되었으나(Plakidas, 1927), 이후 MY18과 D74 분리주(isolate)의 전체게놈이 결정되고 유전자 비교 분석을 통해 Potexvirus로 분류되었고 바이러스 입자의 형태도 480×13 nm의 사상형 입자임이 확인되었다(Jelkmann 등, 1990). 현재까지 SMYEV 전체 게놈 염기서열이 밝혀진 8개 분리주(MY18, D74, Berra 2, AB41-01, AB41-02, NB1165, NS26, AB5-2)에 따르면 SMYEV는 5-6개의 ORF를 발현하며 크기는 5.9-7 kb의 단가닥 RNA 게놈 바이러스임이 구명되었다(Bhagwat 등, 2015; Lamprecht과 Jelkmann, 1997; Ma 등, 2015; Thompson과 Jelkmann, 2004). 유전자 분석은 현재 바이러스의 종 분류에 가장 널리 이용되는 방법으로 Potexvirus의 경우 주로 RNA 중합효소(RNA-dependent RNA polymerase)와 외피단백질(Coat protein, CP) 유전자의 비교분석을 통해 strain이나 variants를 구분한다(King 등, 2012). 유전자 분석에 근거한 계통학적 분석은 바이러스의 진화 및 유전적 변이양상을 파악하는데 매우 중요한 정보가 되고 있다. SMYEV도 계통학적 분석을 통해 유전적으로 확연히 다른 3개의 변이집단이 존재하는 것으로 알려져 있었으나(Thompson과 Jelkmann, 2004), 최근 아르헨티나와 캐나다에서 분리된 SMYEV 분리주에 대한 분석 결과 2개의 새로운 변이집단이 더 존재하는 것이 밝혀져 바이러스 변이주간 다양성이 더 진화되고 있는 것으로 추측된다 (Bhagwat 등, 2015; Ma 등, 2015; Torrico 등, 2016).
본 논문에서는 Cho 등(2011)에 의해 기존에 보고되었던 KNS1 분리주와 최근 국내 육성 딸기 품종에서 분리된 2개의 SMYEV 국내 분리주를 더 확보하여 이들의 CP 유전자 염기서열을 분석하고, 기존 NCBI에 등록된 SMYEV의 48개 분리주들과의 계통학적 분석을 통해 국내 SMYEV 분리주들의 유전적 다양성에 대한 결과를 보고하고자 한다.
재료 및 방법
바이러스 분리주 확보 및 외피단백질 분석
국내 육성 딸기 품종(매향, 설향, 금실, 싼타, 고하, 감홍, 미홍, 장하, 무하)을 대상으로 SMYEV 감염 여부를 조사하였다. 2017년 2월부터 2018년 8월까지 매향, 금실, 설향, 싼타 품종은 경남 진주와 충남 논산 및 부여에서 조사하였고, 고하, 감홍, 미홍, 장하, 무하품종은 강원도 평창 및 고령지농업연구소 내 포장에서 조사하였다. SMYEV 진단은 CP 유전자를 포함하는 특이 프라이머 (SMYEV-FOR: 5’-ACAATCGCCCTGGTCAGTAATTCC-3’, SMYEVREV: 5’-TGGAGCTGGGTAGAAGAAGTGAGA-3’)를 이용하였다. 유전자 진단을 위한 RNA 추출은 TransZol (TransGen Biotech, China)을 이용하였고, SuPrimeScript RT-Premix (GeNet Bio, Korea)로 50°C/30 m; 98°C/30 s; <98°C/10 s, 60°C/30 s, 68°C/1 m> 35 cycles; 68°C/5 m의 조건으로 RT-PCR하였다. PCR 산물은 전기영동에서 확인 후, QIAquick PCR purification kit (Qiagen, Germany)로 정제하여 pGEM-T easy vector(Promega, USA)에 클로닝하고 외피단백질 염기서열을 결정하였다. 그 결과, 설향(약한 모틀)과 감홍(잎 가장자리 변색) 품종에서 SMYEV의 감염을 확인하고 전자현미경으로 사상형 입자를 확인하였다. 각각의 분리주는 SMYEV-SH와 SMYEV-KH으로 명명하고 전체 CP 유전자를 NCBI에 등록하였다.
계통학적 유연관계 및 유전적 다양성 분석
국내에서 분리된 SMYEV의 CP에 대한 계통학적 유전자 분석을 위해 NCBI에 등록된 SMYEV CP 염기서열 정보를 수집하였다(Table 1). 본 연구에서 분리된 SH와 KH, 기존에 분리된 KNS1 국내 분리주를 포함하여 총 48개의 분리주에 대한 계통학적 유연관계를 분석하였다. 염기서열 정렬은 Clustal X2 프로그램을 이용하였고 Phylogenetic tree는 Mega 6의 Neighbor-joining (NJ) 방식으로 작성하였다. NJ-tree의 통계적 유의성은 1000회 반복실행의 bootstrap을 적용하여 분석하였다. Potato virus X의 CP유전자(NC011620)는 outgroup으로 이용하였다. SMYEV의 유전자 다양성 분석을 위해 DnaSP (version 5)을 이용하여 phylogenetic tree에 의해 계통이 분류된 각 subgroup 내 및 subgroup 간 유전거리 및 염기서열 다양성의 요인들(number of monomorphic sites, number of polymorphic site, number of mutations, singleton variable sites, parsimony informative site, synonymous substitutions, nonsynonymous substitutions)을 결정하였다(Librado와 Rozas, 2009).

Information of SMYEV isolates available in the NCBI compared in this study
결과 및 고찰
SMYEV 외피단백질의 국내 분리주 염기서열 분석
국내에서 새로 분리된 SMYEV 2개 분리주 SH와 KH의 CP 유전자의 전체 염기서열을 결정하여 각각 MG418838, MK040457로 GenBank에 등록하였다. SH와 KH 분리주의 CP 전체 염기서열의 길이는 729 bp로 종결코돈을 제외하고 242개의 아미노산을 암호화(coding)하는 것을 확인했다. 분리주 SH와 KH의 CP염기 및 아미노산 서열 상동성은 각각 90.4와 95.5%였다. 기존에 분리된 KNS1을 포함한 국내 분리주 3종의 CP 아미노산 상동성은 95.5-99.8% (Table 2)로, clustal X2 정렬 결과 10곳(4, 20, 22, 31, 40, 67, 72, 74, 112, 158번째 서열)에서 아미노산이 치환되어 있었다(Fig. 1). KNS1는 SH와 같은 품종(설향)에서 분리되었음에도 KH와 더 높은 상동성(99.8%, Table 2)을 보였으며 두 분리주간 치환아미노산은 2곳(31, 40번째)이었다(Fig. 1). 한편, potexvirus 속 바이러스의 CP에서 공통적으로 보존되어 있다고 알려진 염기서열(consensus sequences)인 C 말단의 FAAFDFFDGV (168-174aa residue)서열을 SMYEV 국내 분리주에서도 확인 할 수 있었다(Fig. 1, Bancroft 등, 1991).

Average percent nucleotide sequence and amino acid identities (number in bracket) between the three Korean isolates and the five phylogenetic groups of SMYEV

Alignment of the complete amino acid (aa) sequences of the coat protein of 3 SMYEV Korean isolates. Sequences were aligned by ClustalX2. Consensus aa sequences are located line below. Only those residues differing from consensus are shown for the isolates. Region underlined is the most conserved motifs (FAAFDFF at residue 168-174) shared between SMYEV isolates.
계통유전학적 유연관계 분석
SMYEV의 계통분석에 대한 연구는 2004년에 Thompson과 Jelkmann에 의해 처음 보고 되었는데 24개 분리주에 대한 분석 결과 3개의 subgroup (I, II, III)으로 분류되며, 분리된 기주와 지역에 따라 D74 (subgroup I), 9Redland (subgroup II), MY18 (subgroup III)을 대표 변이주(Type strain)를 지정하였다(Thompson과 Jelkmann, 2004). 그러나 이후 Bhagwat 등(2015)과 Torrico 등(2016)이 각각 캐나다 분리주와 아르헨티나 분리주를 추가하여 각각 계통분석을 한 결과, 5개(I, II, III, IV, V)와 4개(I, II, III, IV)의 subgroup이 새롭게 형성된 phylogenetic tree를 제시한 바 있다(Bhagwat 등, 2015; Torrico 등, 2016). 본 논문에서는 새로 보고하는 2개의 국내 분리주와 현재 NCBI에 등록된 SMYEV 외피단백질에 대한 46개 분리주의 염기서열 정보를 모두 수집하여 총 48개에 대한 계통학적 유연관계를 분석하였다. Neighbor-joining 분석방식에 의한 phylogenetic tree 작성 결과 5개의 subgroup이 형성된 tree를 얻었으며 이 5개 group은 크게 3개의 subgroup (I, II, IV)으로 분리되는 group A 와 2개의 subgroup (III, V)으로 나뉘는 group B로 형성된 tree의 형태였으며 각 group A와 B의 bootstrap value는 71%와 82%였다(Fig. 2). 국내 분리주 KH와 KNS1은 subgroup I에 포함된 반면, SH는 subgroup IV로 분류되어 국내 분리주 간에도 변이가 있음을 알 수 있었다(Fig. 2). Subgroup I은 48개 분리주 중 24개 분리주가 포함되었으며 국내 분리주 2개, 유럽 분리주 12개(독일 8개, 벨기에 1개, 이탈리아 1개, 체코 2개), 아르헨티나 분리주 4개, 미국 분리주 2개, 중국 분리주 2개가 포함되었다. 다양한 지역의 분리주가 한 분류군에 포함되는 것으로 보아 지역간 계통적 유의성은 없는 것으로 사료된다. Subgroup I의 24개 분리주들 간의 염기서열 상동성은 96.3-100%였으며 KH과 KNS1 국내 분리주는 미국분리주인 WSU1988와 98.6%로 가장 가까운 유연관계를 보였다. Subgroup II는 8개 분리주(호주 1개, 아르헨티나 3개, 칠레4개), Subgroup III는 5개 분리주(미국 1개, 아르헨티나 1개, 칠레 3개)가 포함되었고 각 분류군 내 분리주 간 염기서열 상동성은 각각 94.9-98.9%와 97.9-99%였다. 한편, 또 다른 국내 분리주 SH는 캐나다 분리주 3개와 함께 subgroup IV로 분류되었는데 분리주간 염기서열 상동성은 95.6-100% 였으며 그 중 SH와 AB5-2는 염기서열이 100% 일치했다. Subgroup V는 7개 분리주(캐나다 2개, 중국 2개, 아르헨티나 3개)가 포함되었으며 염기서열 상동성은 83.3~99.2%로 가장 낮은 상동성을 보였으나 bootstrap value는 97%로 높게 나타났다.
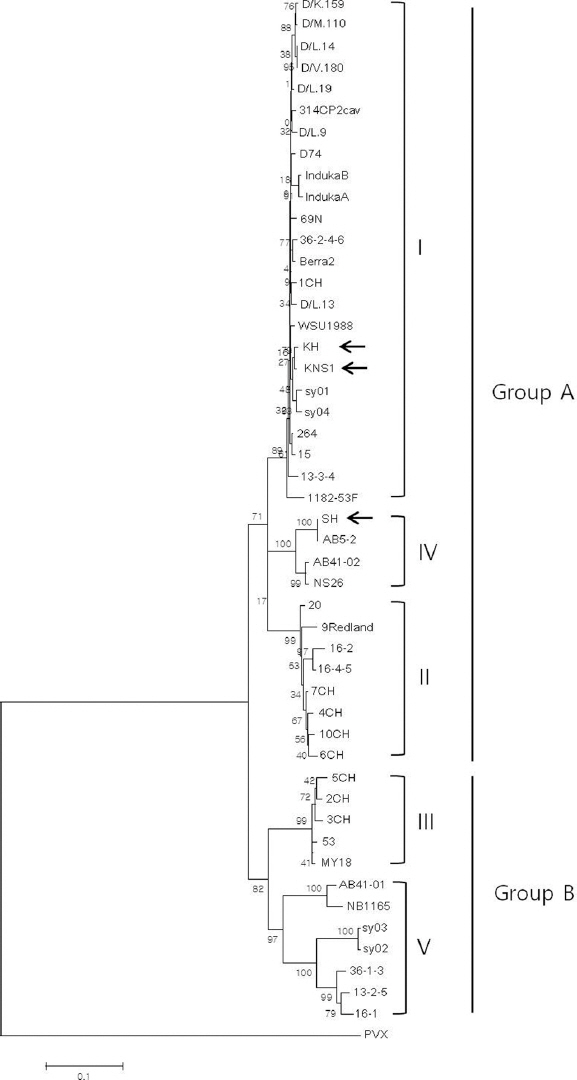
Phylogenetic analysis of the complete nucleotide sequences of the coat protein of different SMYEV isolates (Table 1). Three Korean isolates are marked with arrows. The Potato virus X (PVX; GenBank accession No. NC011620) was included as an outgroup. The phylogenetic trees were reconstructed by the NJ method applying Kimura’s two-parameter method. The numbers on the branches indicate bootstrap percentages based on 1000 replications (only values >60% are shown). The scale bar represents the number of substitutions per base.
5개 subgroup간 유전적 근연관계를 보기 위해 각 subgroup별 5개 대표 변이주(I, D74; II, 9Redland; III, MY18; IV, AB41-02; V, AB41-01)를 선발하고 국내 분리주 3개와 함께 염기와 아미노산서열 상동성을 분석하였다(Table 2). Group A에 포함된 subgroup I과 IV는 염기서열(91.8%)과 아미노산서열(96.7%) 모두 서로 가장 높은 상동성을 보였으며, subgroup II는 I과 염기(91.1%)와 아미노산(96.3%) 서열 상동성이 가장 높았다. Group B에 포함된 subgroup III의 경우, 염기서열 상동성은 88.5%로 IV와 가장 높은 반면 아미노산 서열은 95.1%로 I, II 동일하게 가장 높았다. Subgroup V는 염기(85.7%), 아미노산(94.7%) 서열 모두 III와 가장 높은 상동성을 보였다(Table 2). Phylogenetic tree와 염기서열 상동성 분석 결과, 새로운 2개 subgroup중 IV는 subgroup I과 II에서 파생된 변이 분류군이며 subgroup V는 subgroup III에서 파생된 변이 분류군인 것으로 추측된다. 새로운 변이 분류군 발생에 대한 phylogenetic tree 분석은 SMYEV의 진화 및 다양성을 이해하는 기초자료가 될 것으로 사료된다.
유전적 다양성 분석
SMYEV의 새로운 변이집단이 형성되는 원인은 바이러스가 유전적으로 더 다양하게 진화되고 있음을 의미한다. 특히 RNA 게놈 바이러스는 변이(mutation)와 재조합(recombination)의 비율이 높아 유전자 다양성의 빈도가 높은 것으로 알려져 있다(Garcia-Arenal 등, 2001; Robinson과 Murray, 2013). SMYEV의 경우 phylogenetic tree 분석 결과 새로운 변이주들이 발생되고 있음이 시사되어 각 subgroup에 대한 유전자 다양성 분석을 실시하였다. 각 subgroup의 분리주 간 혹은 집단 간에 나타나는 염기서열 다양성(nucleotide divergence)과 subgroup간 다형성(polymorphism)의 요인들을 분석했다. 각 subgroup (I-V)의 염기서열 다양성은 각각 0.01864(I), 0.02660(II), 0.01838(III), 0.02812(IV), 0.11584(V)로 나타나 subgroup V의 다양성이 가장 높음을 알 수 있었다. 모든 분리주에 대한 염기서열 다양성 수치는 0.0993이였다. Subgroup간 염기서열 다양성은 0.08634-0.18004로 subgroup I과 IV가 가장 낮고, subgroup II와 V가 가장 높았다(Table 3). 다양성 지수는 유전거리를 나타내는 것으로 염기서열의 유사성에 근거하므로 수치가 낮을수록 유전적 유연관계가 높음을 의미한다(Rogers 등, 2012). Subgroup 간 다형성 분석결과는 Table 4에 정리하였다. 유전자 다형성 분석에 가장 큰 요인이 되는 polymorphic site와 parsimony informative site의 비율 분석 결과, 각 subgroup 별 비율은 I: 8.2%와 5.2%; II: 8.9%와 2.1%; III: 4.3%와 0.7%; IV: 4.4%와 3.7%; V: 24.6%와 16.6%로 subgroup V가 가장 높은 다형성을 가지고 있는 것을 확인할 수 있었다. 한편 모든 분석 염기서열에서, 코돈(codon)에는 영향을 주지 않으면서 유전자를 변이시키는 동의치환(synonymous substitution)의 수가 비동의치환(synonymous substitution)에 비해 더 높게 나타났는데 특히 subgroup V의 경우 동의치환의 수가 141로 다른 subgroup보다 훨씬 높았다(Table 4). 국내 분리주의 경우 subgroup I과 IV에만 포함되지만 새로운 변이주가 출현되는지 지속적인 연구가 필요할 것으로 여겨진다. SMYEV의 유전자 다양성에 따른 변이주들의 바이러스 병원성 및 기주범위, 딸기 품종간 저항성 차이 등은 앞으로 더 연구가 진행되어야 할 것으로 사료된다.
Nucleotide divergence of CP gene within and between subgroups of all SMYEV isolates
Information about the polymorphism of SMYEV isolate subgroups
요 약
국내육성딸기 품종인 설향과 감홍에서 딸기누 른오갈바이러스의 국내 분리주 2종을 분리하고 외피단백질 전체 염기서열을 결정하고 분석하였다. 국내 분리주 SH와 KH의 외피단백질 염기와 아미노산 상동성은 각각 90.4%와 95.5% 였다. 기존에 국내에서 보고된 KNS1분리주와 GenBank에 등록된 45개의 다른나라 분리주 외피단백질 염기서열을 모두 수집하여 총 48개 SMYEV 외피단백질에 대한 계통학적 유연관계를 분석할 결과 총 5개의 subgroup (I-V)으로 분류가 되었다. 이 중 subgroup IV과 V과 새로운 변이집단으로 국내분리주도 KH와 KNS1은 subgroup I에 포함된 반면, SH는 새로운 subgroup인 IV에 포함되어 국내분리주간에도 계통이 다른 것을 추측할 수 있었다. 유전적 다양성 분석결과 SMYEV의 새로운 subgroup의 다양성이 더욱 높은 것으로 나타나 SMYEV가 유전적으로 진화를 하고 있음을 알 수 있었다. 이 논문은 국내 SMYEV 분리주에 대한 분자적 특성에 대한 첫 보고이다.
Acknowledgement
This work was supported by a grant from the Agenda Program (PJ01186302) funded by the Rural Development Administration of Korea.
References
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.